
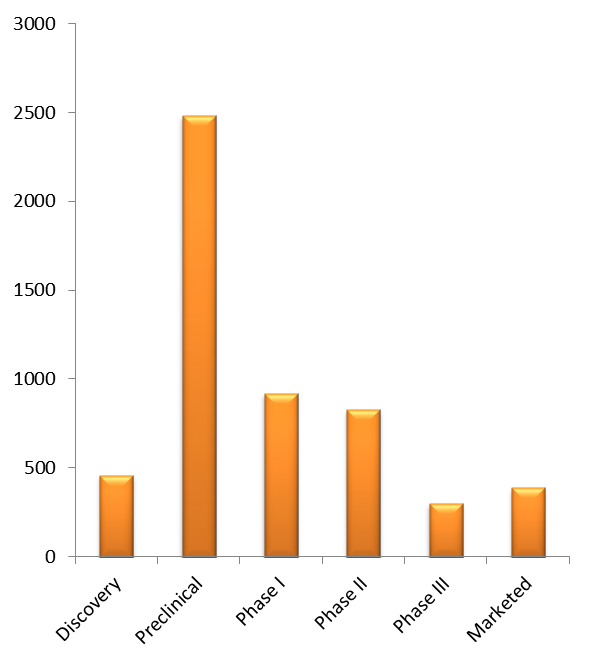
With a pedigree stretching back almost a quarter of a century, BIO-Europe is not only Europe’s largest partnering conference but, according to a recent analysis by 1stOncology™ (also covering BIO International and BIO Asia), it is also one of the world’s richest displays of cancer drug development companies under one roof! With over 600 oncology companies from more than thirty different countries present at BIO-Europe 2018, this is truly a global event. Many of these have also just presented their latest scientific/clinical advancements at the freshly completed European Society of Medical Oncology (ESMO) (Free ESMO Whitepaper) (ESMO 2018) congress. Now coming together at the BIO-Europe 2018 meeting they represent more than 5,500 cancer drugs, from discovery to marketed, and are responsible for more than 40% of the world’s current output in cancer R&D, see pipeline breakdown below.
These new cancer drug technologies are being developed from a wide array of organizations, from centuries old universities such as Jagiellonian University (Poland) founded in 1364, to startup companies like Cedilla Therapeutics (USA) and Epigene Therapeutics (Canada), both founded in 2018. Regardless of age they are all coming together at BIO-Europe 2018 to engage with global life science partners.
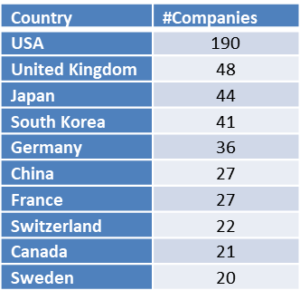
The global nature of this meeting is revealed when looking at the top nations with cancer drug developing companies present at this year BIO-Europe. It is no surprise to see the United States in first place with 190 companies, UK in second with 48 and Japan is in third place with 41 companies. Please see below table for the top ten nations at BIO-Europe 2018.
In today’s fast moving climate where a company can go from an idea to a public company in a blink of an eye, roughly one third of the 600 cancer companies are publicly traded at various stock exchange markets around the world. No less than seven of these have gone through their initial public offering in 2018 alone, namely ARMO BioSciences, ASLAN Pharmaceuticals, Autolus, BeiGene, Forty Seven, MorphoSys and Sutro Biopharma.
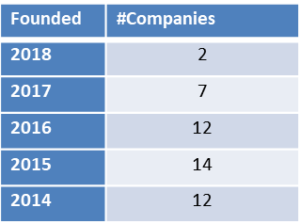
The number of cancer startups, founded in the last five years, present at the BIO-Europe meetings amount to almost fifty, see table below for breakdown per year.
The sizeable cancer pipeline of more than five and a half thousand drugs represented at BIO-Europe is a based on a very diverse selection of technologies and discoveries in cancer biology. Almost one third of these are Immune-Oncology (I-O) drugs including Immune Checkpoint drugs, Cancer vaccines, Bispecific immunomodulators, CAR/TCR therapies and Oncolytic virotherapies, see breakdown by type of I-O drugs below.

In the spotlight of this year’s Nobel Prize in Physiology or Medicine, companies at BIO-Europe feature nearly 300 different immune checkpoint drugs. Other hot progress areas in cancer therapeutics include DNA Damage Response (DDR) drugs, epigenetic therapies, Protein Kinase Inhibitors (PKIs) and Antibody-Dug Conjugates (ADCs).
Schedule your 30 min Free 1stOncology Demo!
Discover why more than 1,500 members use 1stOncology™ to excel in:
Early/Late Stage Pipeline Development - Target Scouting - Clinical Biomarkers - Indication Selection & Expansion - BD&L Contacts - Conference Reports - Combinatorial Drug Settings - Companion Diagnostics - Drug Repositioning - First-in-class Analysis - Competitive Analysis - Deals & Licensing
Schedule Your 30 min Free Demo!