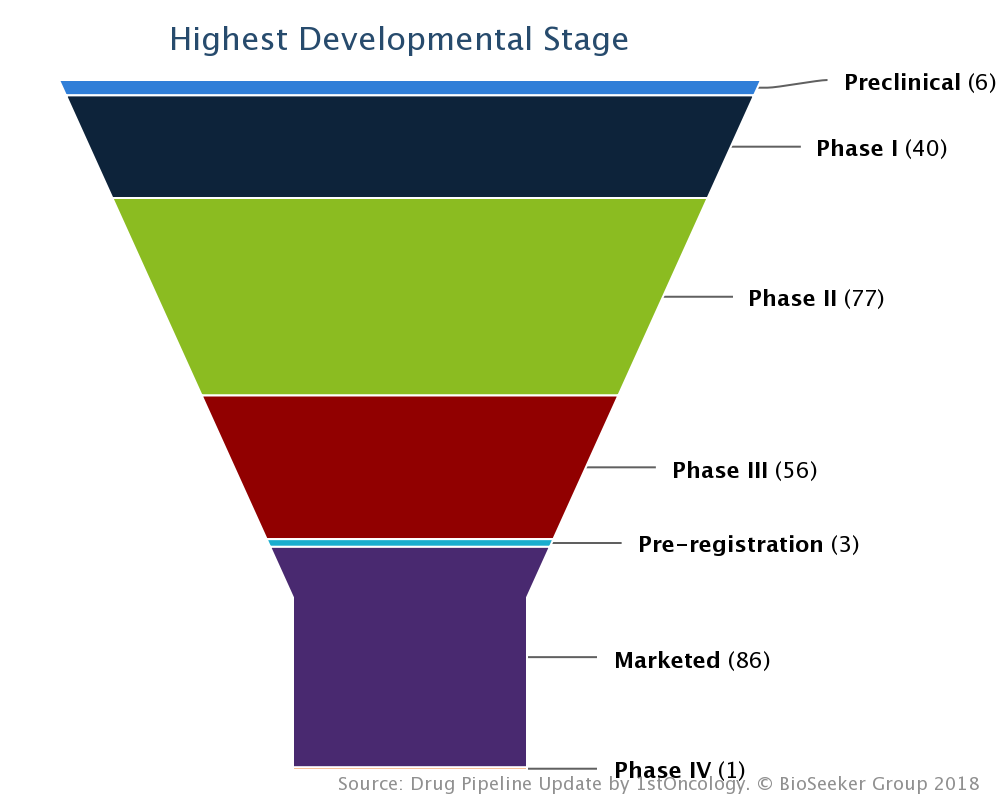
On October 19, 2018 Incyte (NASDAQ:INCY) reported Phase 2 preliminary results of the GEOMETRY mono-1 clinical trial of investigational MET inhibitor capmatinib in 94 adult patients with advanced non-small cell lung cancer (NSCLC) harboring MET exon-14 skipping mutations (Press release, Incyte, OCT 19, 2018, View Source [SID1234530093]). The GEOMETRY mono-1 study showed an overall response rate (ORR) of 72.0 percent (95% CI: 50.6-87.9) in treatment-naive patients and 39.1 percent (95% CI: 27.6-51.6) in previously treated patients. ORR was assessed by blinded independent review committee (BIRC). Adverse events (AEs) were consistent with previously reported data and no new safety signals were observed.
Schedule your 30 min Free 1stOncology Demo!
Discover why more than 1,500 members use 1stOncology™ to excel in:
Early/Late Stage Pipeline Development - Target Scouting - Clinical Biomarkers - Indication Selection & Expansion - BD&L Contacts - Conference Reports - Combinatorial Drug Settings - Companion Diagnostics - Drug Repositioning - First-in-class Analysis - Competitive Analysis - Deals & Licensing
Schedule Your 30 min Free Demo!
Results of the Novartis-sponsored Phase 2 study were presented today at the European Society for Medical Oncology (ESMO) (Free ESMO Whitepaper) 2018 Congress (October 19, 2018 at 4:45 p.m. CEST / 10:45 a.m. EDT, Abstract LBA52).1
"These preliminary findings reveal the potential of capmatinib in MET exon-14 skipping mutated NSCLC patients. Compared to the previously treated patient groups, the primary advantage in terms of overall response rate reported in treatment-naive patients highlights the clinical relevance for an earlier diagnostic testing and prompt treatment of this challenging patient population," said Juergen Wolf, M.D., University Hospital Cologne, Germany.
NSCLC is the most common type of lung cancer, impacting more than 2 million people per year.2 Approximately 3-4 percent of all patients with NSCLC have an identified MET mutation.3 Though rare, this mutation is an indicator of especially poor prognosis and there is currently no approved therapy designed to target this mutation.4
"We are very pleased to announce these promising, preliminary results for capmatinib, another investigational medicine invented at Incyte that has the potential to be the first MET-selective targeted agent approved by the FDA," said Steven Stein, M.D., Chief Medical Officer, Incyte. "We are encouraged by the results of this study and the potential for capmatinib to help patients with advanced MET mutated NSCLC, who face a poor prognosis and represent a clear unmet medical need."
About GEOMETRY mono-1
The GEOMETRY mono-1 trial is a multicenter, open-label, Phase 2 study to evaluate the efficacy and safety of single-agent capmatinib (INC280) in adult patients with EGFR wildtype, ALK-negative rearrangement, advanced NSCLC harboring MET amplification and/or mutations. Patients with MET exon-14 skipping were assigned to Cohorts 4 (previously treated patients) or 5B (treatment naive) regardless of MET amplification/gene copy number (centrally confirmed), and received 400 mg capmatinib tablets twice daily. The primary endpoint was ORR based on BIRC assessment per RECIST v1.1. The key secondary endpoint was duration of response (DOR) by BIRC. The GEOMETRY mono-1 study found an ORR in the treatment-naive patients (n=25) of 72.0 percent (95% CI: 50.6-87.9) and an ORR in the previously treated patients (n=69) of 39.1 percent (95% CI: 27.6-51.6). DOR was not reached by the time of analysis, indicating sustainability of response.1,6
The most common treatment-related AEs included peripheral edema, nausea, vomiting and increased blood creatinine levels. Of patients treated with capmatinib, 83.8 percent experienced an AE, with 33.1 percent having grade 3/4 AEs.1,6
About Capmatinib
Capmatinib (INC280) is an investigational, oral and selective MET inhibitor invented at Incyte that was licensed to Novartis in 2009. Under the Agreement, Incyte granted Novartis exclusive development and commercialization worldwide rights to this MET inhibitor compound and certain back-up compounds in all indications. Novartis has stated that it expects to submit a new drug application to the U.S. Food and Drug Administration for capmatinib as a treatment for patients with advanced non-small cell lung cancer (NSCLC) harboring MET amplification and/or mutations in 2019. If capmatinib is successfully developed by Novartis, Incyte may become eligible for over $500 million in future milestones as well as royalties of between 12 percent and 14 percent on global sales by Novartis.